


国家生物信息中心合作开发原核生物快速泛基因组分析方法
近年来,随着高通量测序技术的迅速发展和细菌基因组测序数量的指数级增长,如何在数以万计的基因组中高效识别同源基因,解析基因组多样性,已成为微生物组学与进化生物学研究的核心挑战。泛基因组分析(Pan-genome analysis)作为揭示细菌种群进化规律与功能分化的重要手段,在理解病原体变异、耐药基因传播及生态适应机制等方面发挥着越来越重要的作用。然而,现有分析方法普遍存在计算效率低、聚类准确率不足及结果定量能力有限等问题,难以支撑当前规模的基因组数据分析需求。
10月15日,国家生物信息中心与中国科学院计算机网络信息中心合作在Nature Communications发表题为“PGAP2: A Comprehensive Toolkit for Prokaryotic Pan-Genome Analysis Based on Fine-grained Feature Networks”的研究论文。该研究提出了新一代原核泛基因组分析工具 PGAP2,通过精炼特征网络(fine-grained feature networks)与双层区域约束聚类策略(dual-level regional restriction strategy),显著提升了聚类精度与运算效率,为大规模原核生物群体基因组研究提供了重要支撑。
研究团队首先基于模拟数据集系统评估了 PGAP2 的聚类精度与稳定性。结果显示,在 16 组不同同源阈值组合的梯度数据集中,PGAP2 的平均聚类准确率(Adjusted Rand Index, ARI)达到 99.97%,显著优于现有工具,且在高多样性组合下仍保持稳定表现。与此同时,PGAP2 在千基因组规模的数据上仅需 6 分钟即可完成完整泛基因组推断,运行速度较Roary提升约 23 倍,内存占用仅为其三分之一。即便在包含超过 4 万株Escherichia coli基因组的超大规模测试中,PGAP2 仍能在合理硬件资源下稳定运行,展现出优异的算法鲁棒性与计算效率。
同时,为了系统评估PGAP2在真实生物数据中的性能差异,研究团队构建了一个覆盖八个主要谱系E. coli金标准泛基因组数据集。该数据集经过人工审编,综合蛋白功能注释、序列相似性、共线性信息及系统发育关系等多维特征构建而成,能够真实反映基因家族的进化谱系。与原始自动聚类结果相比,该金标准数据集显著减少了错误拆分与错误合并事件,并建立了严格定义的同源与旁系同源参考框架。基于该高质量基准数据的对比评估表明,PGAP2 在核心基因(Core)、壳层基因(Shell)及云层基因(Cloud)三类簇中均取得最高准确率,且在复杂旁系同源识别中明显优于其他方法,验证了其算法设计的科学性与泛化能力。
研究团队进一步以人畜共患病原体 Streptococcus suis 的 2,794 个基因组为例,展示了 PGAP2 在复杂多样群体中的应用潜力。PGAP2 在 20 分钟内完成分析,识别出 17,646 个同源基因簇,系统揭示了S. suis基因组的开放式泛基因组结构及其环境适应相关的高变异基因群,为病原体进化与功能研究提供了重要的计算工具。
综上,PGAP2 是一个高精度、高性能、可扩展的原核生物泛基因组分析综合工具,可实现从数据质控、同源基因推断到结果可视化的一站式分析流程。该软件的推广将有助于推动大规模微生物群体基因组学研究的标准化发展,为病原体进化、生态适应及功能解析提供坚实的数据与算法基础。PGAP2源代码可在国家生物信息中心直接下载。
该研究由国家生物信息中心数据资源部肖景发研究员与中国科学院计算机网络信息中心金钟研究员联合攻关团队共同完成。国家生物信息中心肖景发研究员与中国科学院计算机网络信息中心金钟研究员为论文共同通讯作者,国家生物信息中心卜琮凡工程师、博士生张好、中国科学院计算机网络信息中心张凤年博士生为论文共同第一作者。该研究得到国家重点研发计划、中国科学院战略性先导专项、国家自然科学基金等项目资助。
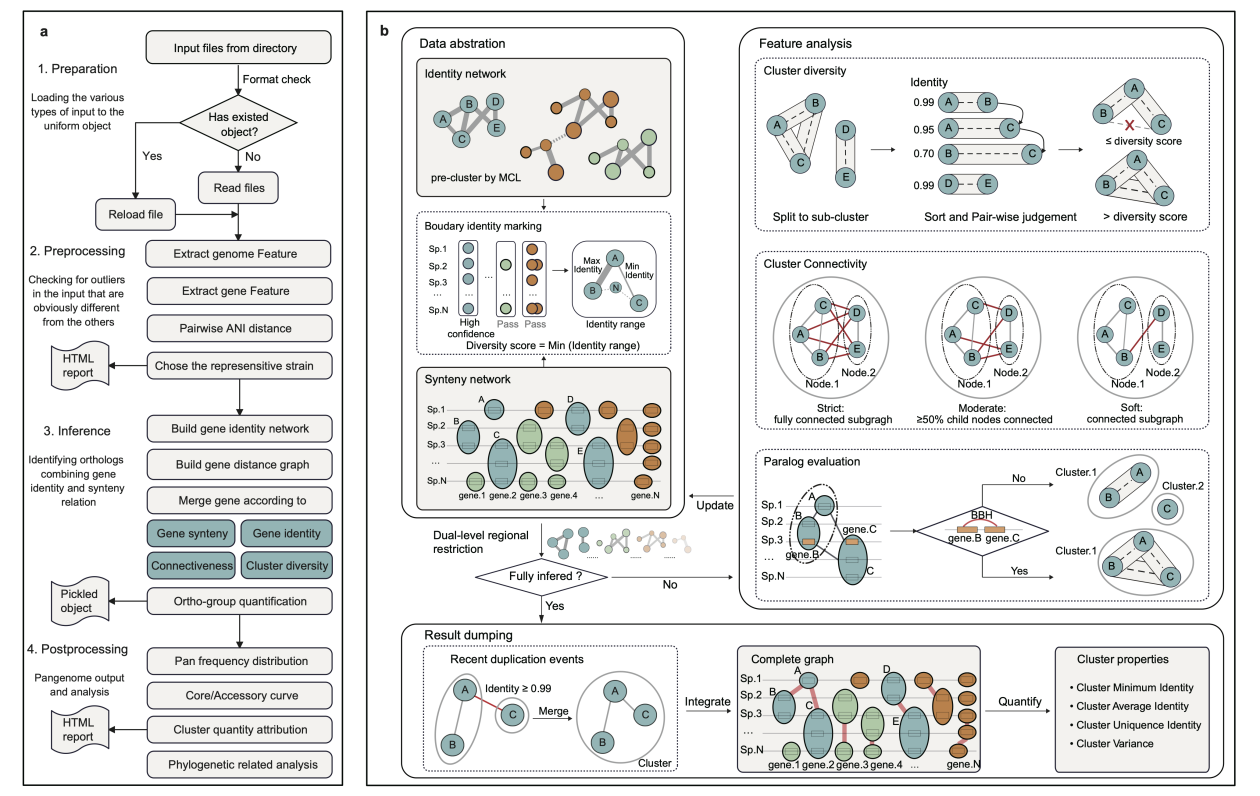
PGAP2方法原理


