
北京基因组所(国家生物信息中心)合作发表多表型全基因组集成分析新方法
全基因组关联研究(Genome-wide association study, GWAS)是研究人类复杂表型遗传因素的有效方法。科学家们已应用GWAS发现了大量的遗传易感位点,阐明了人类复杂表型的多基因性本征,完善了精准医学的核心理论,构建了数千种疾病的遗传风险评估模型,并为多项临床转化类研究提供了明确的分子靶标。然而,由于无法同时分析多个表型,标准的GWAS流程不能高效检出具有多效性的遗传变异。随着人类复杂表型研究体量的与日俱增,如何高效集成分析大量表型亟待解决。
相较于使用个体数据的多表型集成分析方法,基于GWAS汇总数据的方法在多队列合作研究中不受个体数据分享限制,同时受队列特异性影响也更小,因此应用范围更广,但也面临统计学和计算机科学领域的多项挑战。例如需要推导对多表型效应高度敏感的统计量,考虑由遗传和非遗传因素导致的表型间复杂关联关系,校正由多重迭代和样本重叠等因素导致的统计量膨胀,同时在分析大量GWAS的情况下,还要对算法的复杂度、并行化方案和内存占用进行优化。
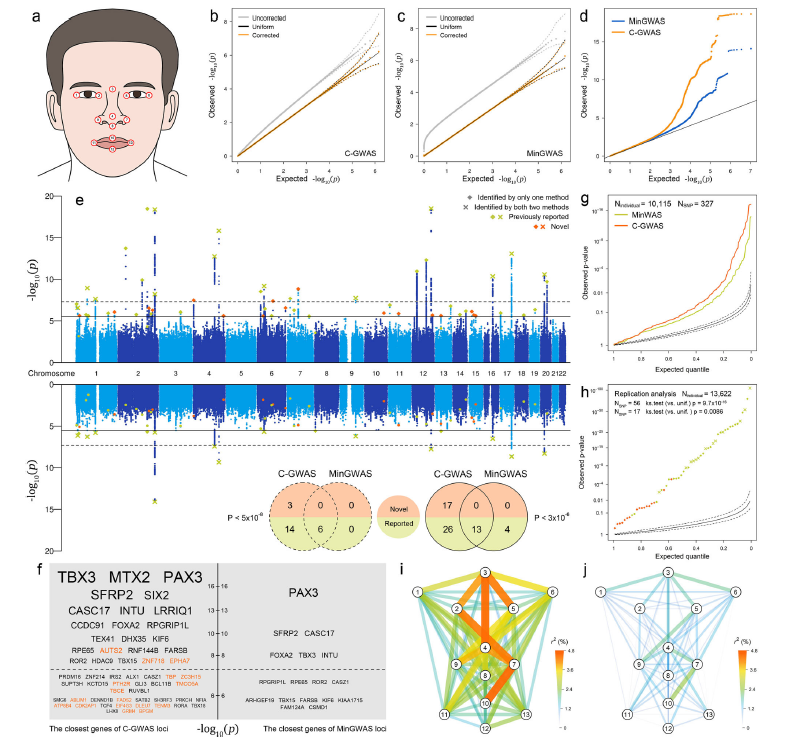
2022年12月20日,北京基因组所(国家生物信息中心)原刘凡研究组与荷兰伊拉斯谟大学Manfred Kayser团队合作在Nature Communications上发表了题为“Combining Genome-wide Association Studies Highlights Novel Loci Involved in Human Facial Variation”的文章,该研究研发了用于集成分析多GWAS的高效算法C-GWAS(Combine GWAS),提供了高度并行优化的开源R软件包,同时通过大规模计算机模拟,展示了C-GWAS对遗传多效性的高检出率和在不同遗传结构下的高稳定性,进而应用C-GWAS分析了78个人类面部形态表型,新发现并验证了一批影响面部形态的遗传变异和功能性基因,加深了对人类多维复杂表型遗传结构的理解。
在方法设计层面,作者对多维GWAS统计量构成的相关性矩阵进行了分解,来区分由可解释遗传因素导致的 “效应相关性” 和由不可解释与非遗传因素导致的 “背景相关性”,进而依据效应和背景相关性相对强度优选合适的统计量进行集成分析,同时引入自适应迭代算法以甄别部分遗传变异仅对特定表型子集有效应的情况,从而实现对遗传多效性检出率的最大化。为了克服由于多重迭代优选引起的统计量膨胀,作者通过计算机模拟获得统计量在零假设下的真实分布,并利用其与均一分布的对应关系,对最终观测到的统计量进行校正,确保了C-GWAS结果和标准GWAS结果可直接在相同显著阈值下进行比较。通过大规模数据模拟发现,与多种其它方法相比,C-GWAS在不同复杂场景下的统计功效和稳定性均展示出明显提升。
人类面部形态代表了一组多维、可遗传且相互关联的复杂表型。作者应用C-GWAS集成分析了78个面部形态的GWAS,结果显示C-GWAS的检出率是传统方法的3倍,并发现了17个影响脸型的新遗传位点。通过进一步的验证分析和功能基因组学分析,作者展示了C-GWAS的结果比传统方法的结果具有更高的遗传多效性,显著提升了脸型可被遗传因素解释的比例,且所指向的靶基因具有更明确的生物发育学功能,表明了C-GWAS在解析多维复杂表型遗传结构中的优势。
在新发现的17个影响面部形态的遗传位点中,有13个位点位于颅神经嵴细胞(Cranial neural crest cells, CNCC)中活性调控元件附近,或在垂体等多个组织中表现出与基因表达eQTL信号的高度共定位。其中,与面部宽度和长度相关的CDK2AP1内含子中的多态性rs10773002,其附近的调控元件在CNCC中调控CDK2AP1的表达,且该位点在多组织中与CDK2AP1的eQTL高度共定位,CDK2AP1编码的蛋白在细胞周期、胚胎干细胞分化和表观遗传调控中发挥作用。这些证据提示该位点通过调控CDK2AP1的表达参与到面部形态形成的过程中。
综上,C-GWAS是一种不依赖个体数据对多表型GWAS汇总数据集成分析的高效算法,对遗传多效性有较高的检出率并在复杂场景下有很强的稳定性。作者提供了高度并行优化的开源R包,可在数小时内集成分析数百个GWAS汇总数据。C-GWAS在人类面部形态数据上的应用成功发现了一批新位点和功能基因,加深了对人类面部形态的遗传结构的理解。未来C-GWAS将被用于解析更高维复杂表型的遗传结构,为多表型间共享遗传因素网络的描绘提供技术支持。