


北京基因组所(国家生物信息中心)合作发现罕见遗传性血液病HLH的新致病基因
噬血细胞综合征(HLH)是一种严重威胁生命的罕见血液病,发病率仅百万分之一,因患者骨髓中常见吞噬血细胞现象而命名。HLH本质是由先天或后天因素导致的免疫系统过度激活,分泌过量细胞因子形成细胞因子风暴,短时间导致多器官功能衰竭,从而危及生命。HLH的病因及发病机制十分复杂,遗传因素被认为是其发病的主要原因之一。依照HLH国际诊疗指南,通过基因检测确认携带HLH易感基因突变的患者即可诊断为原发性HLH,造血干细胞移植是其唯一的治愈手段。目前用于临床筛查的HLH易感基因有12个(PRF1等)。然而,约80%的病人检测不到这些HLH基因突变,其中却有相当一部分呈现家族聚集性、病情反复等原发HLH的典型特征。这些患者很可能存在尚未发现的遗传学异常而延误治疗导致预后不良。因此,在已发现的12个HLH基因之外,是否还存在未知的致病遗传突变?这些突变导致HLH的分子发病机制又是什么?
中国科学院北京基因组研究所(国家生物信息中心)王前飞、刘欣团队与首都医科大学附属北京儿童医院张蕊团队合作,聚焦上述临床问题,开展HLH新致病遗传变异研究,结合家系模型、基因组突变、病人临床表征及分子生物学功能验证,突破性地发现了有助于临床实际应用的HLH新致病基因。研究成果以“NBAS, a gene involved in cytotoxic degranulation, is recurrently mutated in pediatric hemophagocytic lymphohistiocytosis”为题,于2022年7月28日发表在Journal of Hematology & Oncology期刊。
研究团队以未携带已知致病突变的高度疑诊原发HLH病人为研究对象,对13例患儿及其父母进行全基因组或全外显子组测序,通过生物信息学分析将潜在致病因素锁定至病人中重复出现突变的NBAS基因。研究人员进一步在中国HLH队列(224例散发HLH患儿)中验证了NBAS基因突变的重现性,发现其突变发生率(2.11%)仅低于最常见的HLH突变基因PRF1(3.80%)。通过病人原代细胞表型以及体外细胞系功能实验,证实了NBAS基因缺陷可能通过引起NK等免疫细胞的细胞毒功能失调产生HLH。
该研究基于大样本中国HLH患病人群,结合家系模型对先天遗传缺陷明显的HLH患儿进行深入研究,在国际上首次发现HLH的全新致病基因NBAS,并结合功能实验揭示了该基因在HLH中的内在分子机制。这项研究成果拓展了对HLH发病机制的理解,NBAS有望成为第13个HLH临床诊断基因,最终将有助于临床筛查HLH高危人群,提高病人的早诊早治率。
中国科学院北京基因组研究所(国家生物信息中心)王前飞研究员、刘欣研究员以及首都医科大学附属北京儿童医院血液科张蕊主任为本文共同通讯作者,课题组博士毕业生毕小慢、陈蕾和首都医科大学附属北京儿童医院副研究员张晴为本文的共同第一作者。该研究得到了国家重点研发计划、国家自然科学基金、北京市自然科学基金等项目资助。
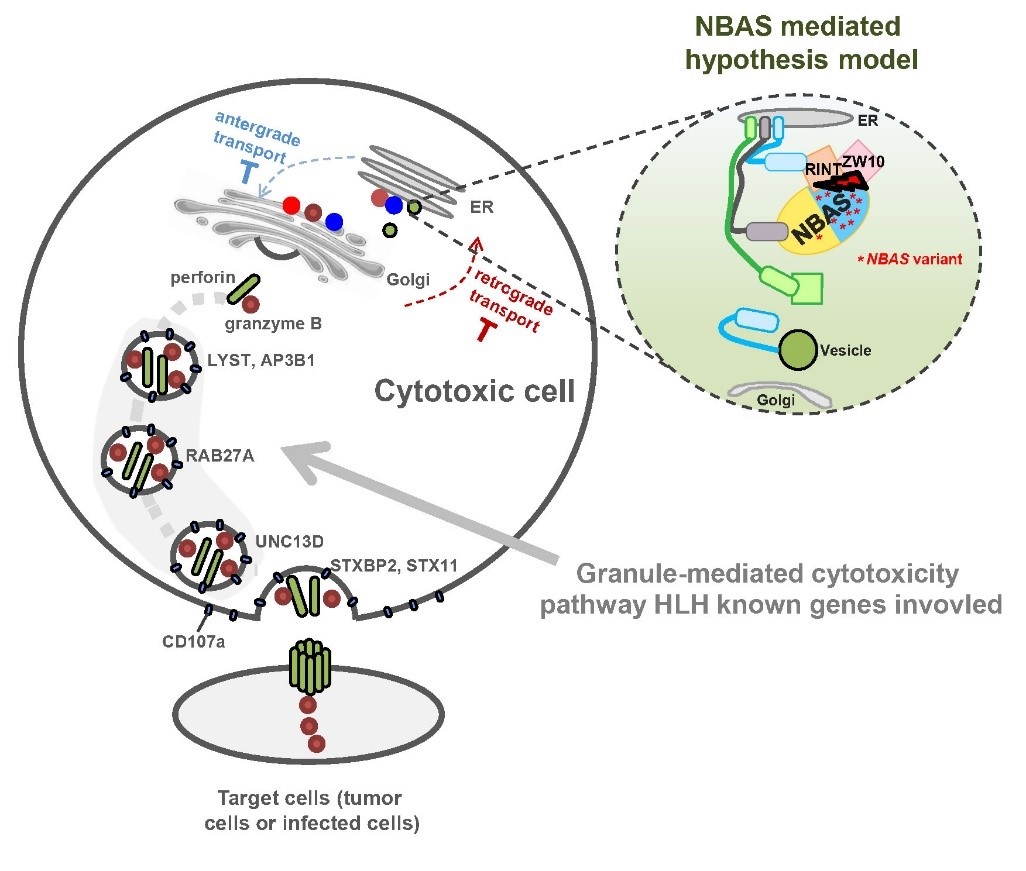
NBAS基因缺陷引起HLH发生的假说模型


