


国家生物信息中心开发针对长读长宏转录组数据的分析工具Fungen
微生物在维持生态系统的稳定性和功能中扮演着至关重要的角色。细菌和古菌等原核生物的多样性及其功能已经被广泛研究,尽管真菌及其他真核微生物在生态系统中同样发挥着关键作用,但由于缺乏高质量的参考基因组,且其基因功能研究相对滞后,真核微生物的基因多样性和代谢潜力仍未被充分探索。
近年来,长读长测序技术的兴起,为研究真核微生物的转录组提供了新机遇。长读长宏转录组(Long-read metatranscriptomics)测序成为解析复杂环境中真核微生物活性基因的强大工具。然而,该技术在应用过程中仍然面临着两个主要挑战:一是缺乏高质量的参考基因组,二是测序错误率较高。
Fungen:突破长读长宏转录组分析瓶颈的新方法
针对上述挑战,国家生物信息中心高远团队开发了一款名为 Fungen 的创新性软件工具,Fungen是一种针对长读长宏转录组数据的分析方法,能够在不依赖参考基因组的情况下,对长读长测序数据进行基因水平的聚类和测序错误校正,以更高效、更精准的方式解析环境样本中的真核微生物转录本,相关研究近日发表于Science China Life Sciences 期刊。
Fungen的核心优势在于其高效的算法设计,通过结合用于读段聚类的minimizer 3-mer快速匹配和网络数据结构,实现了对宏转录组数据的快速处理。与现有工具相比,Fungen不仅减少了计算资源占用,还在速度上相较现有方法提升了22至56倍。此外,Fungen克服了近缘物种之间同源基因相似性带来的挑战,通过独特的算法设计,能够准确地区分这些相似基因,从而获得高精度的转录本序列。
Fungen在真实环境数据中的应用
为了验证Fungen在真实宏转录组数据中的应用价值,研究团队使用该工具分析了多种环境样本的长读长测序数据,包括宏转录组的模拟数据、真菌的合成宏转录组数据、远洋海水的direct RNA测序数据以及多种环境的全长rRNA扩增子测序数据。结果表明,Fungen不仅能够对测序读段进行可靠的基因水平聚类,还能获得高精度的转录本序列。
值得一提的是,当应用于农田与湿地土壤的高深度 cDNA 测序数据时,Fungen能够在真菌物种水平上重建原位基因表达动态,揭示了植物病原真菌在土壤环境中的特殊生存策略,为理解植物病原真菌的生态行为提供了新的视角。
Fungen的意义与展望
Fungen为分析复杂的宏转录组全长数据提供了快速和准确的解决方案。其无参考基因组的方法不仅适用于海洋、土壤等复杂环境样本的研究,还为揭示真核微生物的多样性和功能提供了关键技术支持。
未来,随着长读长测序技术的不断进步,Fungen可望被应用于更多环境微生物学、生态学和医学研究领域,例如人类肠道微生物组、真菌病原体鉴定、工业微生物代谢研究等,为探索微生物世界的奥秘提供更强大的工具支持。
该研究得到了国家重点研发计划、中国科学院前瞻战略科技先导专项(A类先导专项),北京市杰出青年科学基金等项目资助。
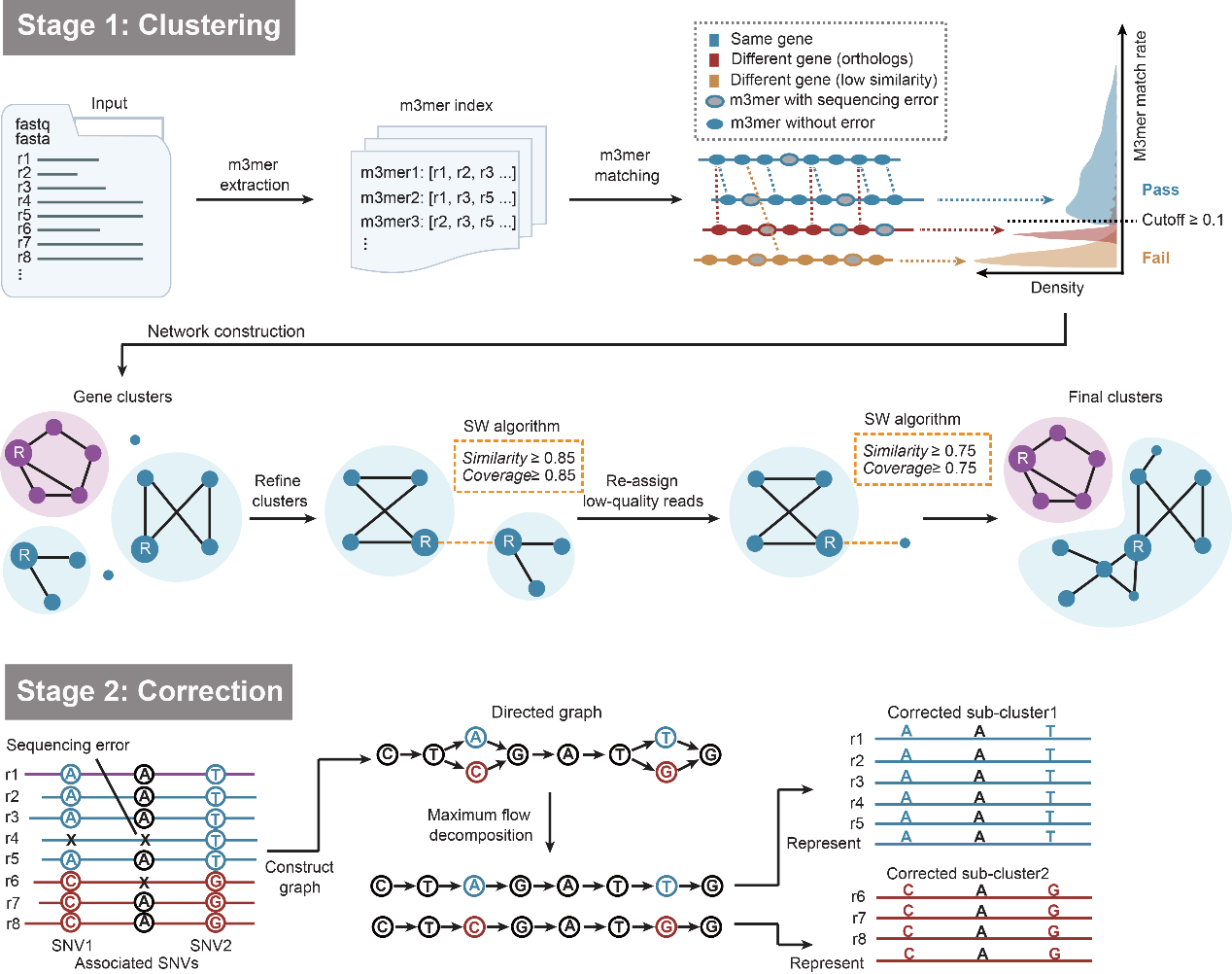
Fungen的核心工作流程


