


北京基因组所揭示骨髓多能干祖细胞在遗传性疾病中的重要作用
近日,中国科学院北京基因组研究所精准基因组医学重点实验室研究员王前飞和美国迈阿密大学米勒医学院杨逢春教授合作,揭示了Bohring-Opitz综合征的胚系新生突变基因ASXL1在骨髓基质多能祖细胞功能和骨骼发育维持中的关键作用,这一重要研究成果发表在Stem Cell Reports杂志。
骨髓基质多能祖细胞(BMSC)是具有自我更新和多谱系分化潜能的多能祖细胞。BMSC中骨生成和脂肪细胞生成的关系对于正常骨骼体内平衡非常关键,这些细胞的分化命运出现倾斜会导致人体发育异常。Bohring-Opitz综合征(Bohring-Opitz syndrome, BOS)是一种导致幼童死亡的异质性遗传病,其特征是严重的发育迟缓、上肢固定挛缩、姿势异常、喂养困难、严重的智力障碍等。以往研究发现,BOS病人存在ASXL1基因的胚系新生突变,导致ASXL1蛋白功能缺失。然而,作为转录激活/抑制关键调节蛋白TrxG和PcG的增强子,ASXL1基因突变引起BOS的细胞和分子机制仍不明确。
研究人员在Asxl1靶向小鼠模型中发现,成骨细胞及其祖细胞的Asxl1基因完全敲除和条件性敲除导致严重的骨骼缺失和骨髓基质多能祖细胞数目减少;Asxl1敲除后的骨髓基质多能祖细胞展现出自我更新能力受损和分化倾向改变,分化为成骨细胞的潜力显著下降,偏向分化为脂肪细胞。转录组测序及其生物信息学分析表明,一些与细胞增殖、骨骼发育和形态发生相关的基因表达发生改变;而且,基因富集分析显示干细胞自我更新的印记基因表达下调,表明Asxl1对调控骨髓基质多能祖细胞的干性有作用。研究人员进一步发现,Asxl1重新表达使干性基因NANOG和OCT4的表达正常化,恢复了Asxl1敲除的骨髓基质多能祖细胞的自我更新能力。
ASXL1基因的体细胞突变发生在髓系恶性疾病中,包括骨髓增生异常综合征(myelodysplastic syndrome)、慢性粒单核细胞白血病(chronic myelomonocytic leukemia)和急性髓系白血病(acute myeloid leukemia)。该研究揭示了ASXL1通过影响骨髓基质多能祖细胞功能和骨骼发育在Bohring-Opitz综合征发病中起重要作用,为代谢性骨骼疾病及癌症的临床治疗提供了新的机遇。
该研究获得了国家自然科学基金委和中科院国际合作局对外合作重点项目的资助。
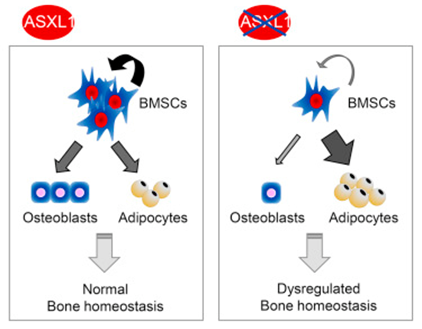
Bohring-Opitz综合征胚系突变基因ASXL1在骨髓基质多能祖细胞功能和骨骼发育维持中起关键作用


