在过去的几十年中,人们往往使用高度保守的基因家族进行系统进化分析,采用全基因组序列进行系统进化分析并不普遍。目前,基于是否进行序列的比对,分子系统发生树的构建分为两类。其中,不需要进行序列比对的方法是依据K-mer向量计算的距离矩阵进行系统进化分析,大量的研究证实该算法是行之有效的,尤其是对基因组中诸如蛋白编码序列等的特定区域。不仅如此,K-mer算法还在组学的其他方面,包括基因组组装、motif预测、重复序列的识别以及基因组的复杂性评估等都受到了广泛的关注。基于K-mer算法在组学中的重要表现,在这个大规模基因组数据快速积累的时代,构建一个基于K-mer算法易于存储并且将大量基因组数据可视化处理的数据库十分迫切。
为此,中国科学院北京基因组研究所基因组科学与信息重点实验室于军组和英国伦敦大学学院(UCL)肿瘤研究所王大鹏博士合作开发了一套基于K-mer算法的基因组组分分析数据库KGCAK。此项研究于近期发表在Biology Direct杂志。
在这个数据库中,研究人员搜集了Ensembl、Phytozome和NCBI等几大主流基因组数据库中包括高等动植物、原生生物、真菌、细菌、病毒等在内的8000多个核基因组或者细胞器基因组,同时包括基因组不同维度的序列,主要有DNA、cDNA、CDS、氨基酸和ncRNA序列。并且还分别计算和存储了核酸序列(K从2变化到10)和氨基酸序列(K从1变化到5)的K-mer向量,以方便进行不同维度数据跨物种的系统发生树构建。此外,该数据库提供了评估不同物种基因组复杂度的交互工具,主要包括基因组基本特征参数、K-mer向量的数学参数统计、频率分布、唯一性比率,以及二维和三维空间可视化分析基因组参数和K-mer参数的交互关系等。
总的来说,该数据库通过捕获基因组序列特征并把基因组转化成更易于理解和可视化的数字K-mer向量,以期通过K-mer算法用可视化的图形和定量的数据构建一个比较基因组学的平台,将为系统发生树构建和通过基因组数据研究物种关系提供良好的参照和指引。
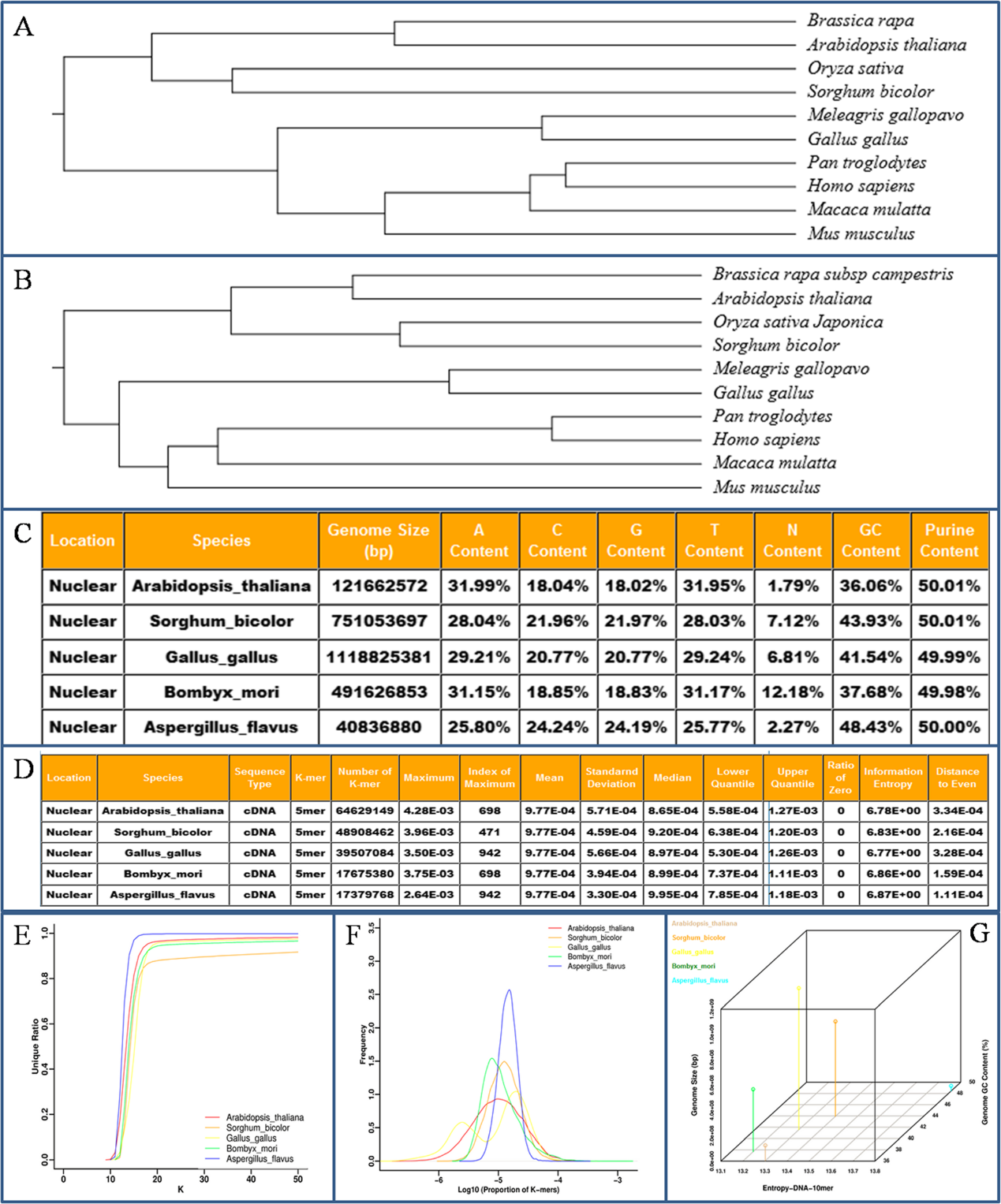
KGCAK数据库中基本功能模块举例


